![]()
XVII. METHYLAMINE
The following information was obtained from Dr. Helmut Hanishch on 12th and 14th May. Reference should be made to Figure XXXVII.
About 80 T/month of mono- and di-methylamine were made. A batch mixture was made up, presumably at pressure above atmospheric, containing 4 to 5 mols NH3 to 1 mol CH3OH, and the mixture was pumped at the rate of about 1000 1/hr. to a converter. This was run at any pressure from 60 to 200ats, but the effect of pressure was not ascertained. The converter and interchanger were both made of S1 steel (low carbon steel) with copper lining and the electrical preheater was copper-covered. The converter was 500 mms I.D. and 8000 mms long, containing about 900 1. of catalyst. There was said to be little difference in performance between catalyst 6069 (90% Al2O2 and be little difference in performance between catalyst 6069 (90% Al2O3 and 10% kaolin) and catalyst 6067 (50% Al2O3 and 50% kaolin), samples of which were obtained.
The make was let-down into a reservoir kept at 25 ats. The de-watering still was copper-lined and filled with 18 m of porcelain Raschig rings, run at a pressure of 20 ats.
Trimethylamine (TMA) and excess NH3 were then separated from the mono- and di (MMS and DMA) in a continuous double still run at 15 ats. The top column contained bubble-plates, with a TMA-NH3 azeotrope taken overhead and NH3 taken from the bottom. The bottom column was filled with Raschig ringswith crude MMA and DMA mixture taken off the bottom.
The TMA-NH3 azeotrope was fed back continuously into a similar converter, for partial reconversion to MMA and DMA. The product was fed into the some do-watering still as the main steam.
Final purification of the crude MMA and DMA mixture was carried out in a batch still. A batch consisted of 30 M3 of the crude mixture, with 5 M3 NH3 added to provide NH3 for TMA separation. The still had an I.D. of 700 mm and was paced with 18 m of Raschig rings. With the top temperature kept at 40° C and he bottom temperature at 55° to 60°C, the TMA-NH3 azeotrope was taken off as the pressure was dropped from 15 to 10 ats, then MMA as the pressure was dropped further to 8 ats. And then DMA as the pressure was dropped to 5 ats, with water left behind. The MMA and DMA fractions were then given a further final purification.
Dr. Hanisch said that copper could be used in this process if care was taken to exclude oxygen.
ISOBUTYLAMINE MANUFACTURE
Dr. Hanisch said that 20 to 25 T/month of higher alkyl amines were made and gave the following details of the manufacture of isobutylamine as typical.
In contrast to methylamines, which were made from methanol and ammonia over a dehydrating catalyst, isobutylamine was made from the aldehyde and ammonia in the presence of hydrogen.
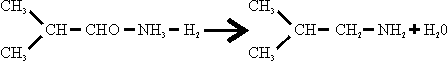
The only details obtained of the manufacture of isobutyraldehyde were that, whereas formerly this had been made by passing isobutanol at 1 at. And 370°C over 2493 (zinc sulphide on pumice, a sample of which was obtained), the method now preferred was to pass isobutanol with air over a silver gauze.
The synthesis of isobutylamine was carried out at 220 ats. Over catalyst 3076, NiS.WS2, a sample of which was obtained, at a temperature of 300° C. 1800 to 2000 m3/hr. H2 and 600 to 800 1/hr. NH3 were circulated with a feed of 601/hr. isobutyraldehyde over 90 to 100 1 catalyst. The product was separated after cooling and the surplus H2 recirculated. The crude was stored at 50 ats. And distilled for purification, releasing surplus NH3, which was recirculated.
Amines of other higher alcohols were made in a similar way.
SCHIFF’S BASE
Dr. Hanisch said this was made by reacting isobutylamine and isobutyraldehyde at 1 at (the catalyst, if any, was not specified and hydrogenating the product at 200-220 ats.
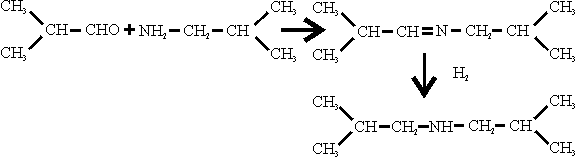
The hydrogenation was carried out in apparatus similar to that used for the manufacture of isobutylamine, using the same catalyst, NiS.WS2. The feed rate at Schiff’s base was 40 1/hr. and the make-up H2 rate was 40 to 50 M3/hr, the temperature being 220 to 310°C; the catalyst volume was only 40 l.