![]()
THE SYNTHESIS OF HYDROCARBONS AND CHEMICALS FROM CO AND H2
SECTION VII
THE OXO-SYNTHESIS
SUMMARY
The attached report covers the development of a new synthesis of higher boiling alcohols by reacting olefins with CO and H2 over cobalt and pilot plant stage and resulted in the erection of one 10,000 ton/year plant by Ruhrchemie AG at Holten.
1. General Introduction. (see ref. VII/1 and VII/2 at the end of this section).
Oxo-synthesis is a process for the production of alcohols by the reaction of olefins with one molecule each of CO and H2 and subsequent hydrogenation of the resulting aldehyde.
The process was developed independently by Ruhrchemie and I.G. Farben. During the war these companies arrived at an agreement whereby they would pool their information, but up to this time only one commercial plant has been erected (by “oxogesellschaft”m.b.H. at Holten). The unit was never started and thus no actual plant performance data are available. All information given below is based on large scale pilot plant operation and laboratory work.
The Ruhrchemie plant is based on a batch type operation, while I.G. Leuna had developed a continuous sump-phase type process. It is felt that the I.G. process is ready to be put into commercial practice and is superior to RCH. The main improvement consists in the continuous operation and somewhat high output per catalyst volume.
The principles involved and their practical application are described below:
2. Chemistry of the Oxo-Synthesis. (See ref. VII/3, VII/5 to VII/7 at end of this sect.)
The following reactions occur when olefins are contacted with CO-H2 mixtures over certain catalyst:
(1) Formation of aldehyde
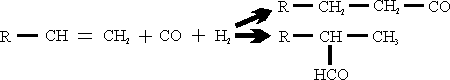
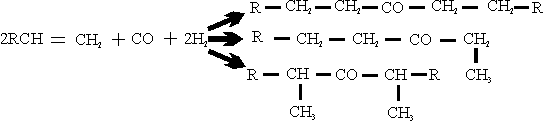
A great deal of work has been done to study these reactions and some of the results are listed below:
Pure olefins of different types were subjected to the Oxo reaction and the products separated and each chemical individual analyzed in detail.
| For example: | pure 2 methylpentene - 1 (Isohexane) |
| pure dodecylene - 1 | |
| pure cyclohexene |
were reacted with CO+H2. The catalyst was removed and the CO-carbonyl decomposed by washing with 5% H2SO4. The products were then analyzed. (Note: that this is only the first step of the oxo-synthesis). The aldehydes are not easily analyzed since they are highly reactive. It is known that more alcohols are usually formed in the oxo synthesis than would be expected from the analytical determination of the aldehydes after the first step.
(c) Products from 2 Methylpentene - 1
45% of product boiling below 200° C (760mm.) identified in this fraction were:
3 methyl hexylaldehyde (main product)
3 methyl hexylol
3 methyl capronic acid
In the higher boiling fraction the following products could be found:
2, 8 dimetheyl-undecanone - 6
Higher ester of 2-methylcapronic acid.
These products correspond exactly to the basic reactions listed at the beginning of this chapter.
In parallel fashion cyclohexene was treated and the following identified in the products:
|
hexahydro- benzaldehyde |
|
dimmer of benzaldehyde |
|
trimer of benzaldehyde |
|
hexahydro benzyl alcohol |
|
hexahydro benzoic acid - hexahydro benzyl - ester. |
The compounds charged to Oxosynthesis, particularly those obtained from F.T. type operations are known to be substantially terminal olefins. Yet it was found that some of the products could have been formed only if a double bond shift had preceded the formation of the aldehyde. This effect was studied and the results are summarized below:
Again n-dodecylene - 1 was used as starting material, and subjected to Oxo synthesis. But the reaction was carried through to the alcohol. In order then to establish the structure of the alcohols, they were first carefully dehydrated and the olefine was next split by oxidation and treatment with Ag2O. The resulting acids were checked for their chain length and thus the branching was determined, which could only be an effect of a shift of the double bond of the n-dodecylene - 1 feed during the Oxo-reaction.
These tests proved, that from terminal normal olefins the oxosynthesis yields branched alcohols, in particular 2-alkyl alcohols, whereby the yield decreases with increasing length of the sidechain.
It was finally possible to prove that cobalt carbonyl was the catalyst responsible for the double bond shift. Dodecylene-1 was treated at 150 to 200° C and 200 atm. of CO and with Co-thoria catalyst and all isomer dodecylenes were found in almost equimolecular ratio.
Fe-carbonyl showed a similar effect, but not the same activity, as only 40 to 45% of the dodecylene - 1 was isomerized. Nickel however, had no effect at all. These findings are interesting because the three named metals catalyze the Oxo reaction in about the same extent as they isomerize the double bond.
The shift of the double bond was made the subject of a patent application by I.G. Farben. (See reference VII/9 at end of this section). The disclosure involves the treatment of olefine hydrocarbons with metalcarbonyl (particularly cobalt-carbonyl) at 70 atm. CO pressure and at 150° C for two hours with 3% catalyst in the feed as an example. The process should serve to raise the octane number of the hydrocarbon. (Note: that similar tests were carried out on synololefines, which were to be used as feed stock for the Oxo-synthesis). The olefins were found to be at least 90% straight chained.
The Synololefines, on the other hand, are not necessarily all terminal olefins. These facts seem to constitute the proof that the Synol reaction (for alcohols) does not consist in an Oxo type synthesis carried out on initially formed olefins. If such were the case, the synol alcohols would have to contain about 50% a-substituted alcohols, which they do not. Much rather it is possible that they synol olefins are the result of a dehydration of the primarily formed terminal alcohol, followed by a shift of the double bond.
That Fe has a tendency to shift the double bond, was seen from the analysis of a Synol fraction (undecylene: 73°-78° C at 10 mm. Hg.) which had been synthesized over the standard iron catalyst:
|
Undecylene |
D1 |
D2 |
D3 |
D4 |
D5 |
D6 |
|
Mol. % |
60 |
27 |
2 |
1 |
0 |
This compares as follows with the distribution of double bond, obtained from n-dedecylene-1 by treatment with Co(CO)4
|
Undecylene |
D1 |
D2 |
D3 |
D4 |
D5 |
D6 |
|
Mol. % |
8.1 |
27.2 |
23.0 |
18.1 |
13.3 |
10.3 |
3. Olefines for the Oxo Reaction.
The following materials were considered as feed for the oxo-synthesis:
|
Olefines from Fischer oil (Kreislauf operation) |
|
Olefines from Cracked Fischer wax |
|
Olefines from Fe catalyst F.T. operation |
|
Olefines from Synol operation |
|
Olefines from Various cracked mineral oils |
|
Olefines from Shale oils |
|
Olefines from Hydrogenation products. |
It appears that the reaction is applicable in principle to all mono-olefines independent of origin. The limitations probably arise from the impurities contained in the olefins, such as aromatics, gums, or catalyst poisons. It is for this reason, that synthetic olefins are preferred, but good results were obtained from mineral oil olefins in spite of their sulphur content. The temperature required for the synthesis is somewhat higher and the sulphur content is a drawback in the hydrogenation stage, unless it is carried out quite independently over sulphide catalysts.
Olefines containing charging stocks from different synthesis operations were studied extensively, to evaluate their use for the oxo reaction. The following products were tested:
|
Catalyst used in synthesis |
|
|
Ruhrchemie, primary synthesis olefines |
Cobalt |
|
Ruhrchemie, thermally cracked was |
Cobalt |
|
Synol olefins |
Cobalt |
|
Michael (I.G. “Schaumfahrweise”) olefines |
Iron |
|
Lurgi olefins |
Iron |
The conclusions were as follows:
In order to compare the different synthesis fractions for the oxo-synthesis, the following table has been prepared: (See attached sheet)
Special Feed Stocks:
It was attempted to apply to oxo reaction on other compounds containing double bonds, with the following results:
|
Tetra methylautadiene: |
Reaction only with one double bond |
|
Dimethylhexadiene: |
Only 40% of expected product; rest is hydrogenated at one double bond, while other reacts according to Oxo reaction. |
|
Allylalcohol: |
Reaction products very complex. No detailed analysis available. |
The catalyst is identical with the standard cobalt Fischer-Tropsch catalyst. It is applied as a slurry in the liquid feed. Usually in concentration of 3% wt. catalyst based on olefine in the feed. At the end of the operation the catalyst if filtered from the reactants through a ceramic disc or thimble and returned for further use. About 100 batches can be processed with one catalyst charge.
The catalyst contained cobalt, thoria, kiesleguhr in the customary ratio 100-1.5-200 (weight %). It did not contain magnesia, because it was found that MgO encouraged condensation of aldehydes. The catalyst had an apparent density of 0.3 to 0.35. Thus 10 kg. of oil contained about one liter of the catalyst. RCH used batch operation and carefully avoided all pumping of the slurry. The liquid was removed from one vessel to another by gas displacement. In the continuous operation used by I.G. it was found that the kieselguhr caused considerable trouble due to abrasion in the pump valves. Attempts to replace the kieselguhr with talcum failed.
One of the main problems in the process is the formation of cobalt carbonyl. (Its effect on the double bond has already been described). I.G. Leuna developed a cobalt-copper catalyst, which gave only a fraction of the carbonyl (less than 10 milligram/liter). The copper has a stabilizing effect on the cobalt. This catalyst was satisfactory for the 1st stage (aldehyde formation) but did not hydrogenate well.
The activity of the standard catalyst for the 2nd stage (hydrogenation) is considerably diminished by the presence of even small quantities of CO in the hydrogen. This sensitivity becomes more pronounced with the age of the catalyst. The installation of a “Methanizer” was therefore considered to remove this residual - CO from the H2-cycle in the 2nd stage.
TABLE II CONTENT OF OLEFINES AND OXYGENATED COMPOUNDS OF VARIOUS SYNTHESIS PRODUCTS
|
Fraction ° C |
Number of Carbon atoms |
“Kreislauf” middle oil |
RCH therm . cracked- wax |
Synol “Benzin Fahrweise” |
Synol Alcohol Operation |
Michael Synthesis Product |
Lurgi Synthesis Product |
||||||
|
ole- |
Oxygtd Compds |
ole- |
Oxygtd Compds |
ole- |
Oxygtd Compds |
ole- |
Oxygtd Compds |
ole- |
Oxygtd Compds |
ole- |
Oxygtd Compds |
||
|
105-175 |
8-10 |
42 |
16 |
80 |
0 |
- |
- |
17 |
65 |
- |
- |
- |
- |
|
175-218 |
11-12 |
41 |
10 |
80 |
0 |
46 |
20 |
18 |
68 |
46 |
36 |
52 |
7 |
|
218-255 |
13-14 |
33 |
9 |
75 |
0 |
47 |
21 |
16 |
68 |
48 |
25 |
55 |
5 |
|
255-290 |
15-16 |
28 |
9 |
50 |
0 |
43 |
21 |
16 |
68 |
45 |
12 |
50 |
4 |
|
290-320 |
17-18 |
20 |
11 |
40 |
0 |
42 |
20 |
18 |
69 |
40 |
14 |
45 |
7 |
|
320-260 |
- |
15 |
5 |
- |
- |
45 |
18 |
17 |
59 |
32 |
20 |
- |
- |
All values are weight %.
Note: That the Oxygenated compounds include alcohols, aldehydes, acids and esters. For detail breakdown, see attached German report.
See also Reference VII/5 at end of this section.
Another modification of the continuous process had been developed by I.G. Farben at Ludwigshafen. (See also reference VII/17 at end of this section). In this process cobalt acetate is contacted with fatty acids (from Oxo-alcohols) in an autoclave to yield the cobaltsalt of the fatty acid. (The free acetic acid is condensed).
Thus the cobalt is introduced in solution into the feed to give a 0.02-0.05% Co concentration. This mixture is charged to the reactor where it passes over a solid cobalt catalyst bed (Co on pumice). The mixture is finally freed from the cobalt in a special smaller reactor, which is filled with plain pumice. Hydrogen is added here and the reduced cobalt deposited on the pumice. After several months the final reactor must be purged of cobalt. This is done by CO, which forms carbonyl; the carbonyl is again dissolved in oil and returned to the feed.
This process does not require any filtration step. The gas streams of the process must be continuously checked for Co carbonyl. It is necessary to control its decomposition, or if possible, direct it to a part of the plant where it does no harm. In general, it is scrubbed out in a wash column using the olefine feed as sponge. Cobalt deposition throughout the plant due to Co carbonyl has been a considerable problem in the continuous process.
The catalyst, as any F.T. catalyst, must be reduced. In the particular case of the oxo-synthesis, where a slurry is used instead of a solid bed, the pelleting of the catalyst is unnecessary. This however, calls for some special arrangement to reduce the catalyst. It could be pelleted, reduced and again broken up, but the I.G. engineers proposed reduction in “fluid phase”. In a patent application dated 9 July 1942, the use of a funnel shaped vessel was disclosed for the reduction of catalyst powder, (See reference VII/8 at end of this section) where the reducing gas would keep the dust in motion. A filter or other means could be used to remove the catalyst from the gas stream which was recycled through the catalyst.